单一产物选择性是催化反应工业应用的主要挑战和亟需解决的关键科学问题。高选择性地氧化伯醇到羧酸在大宗化学品制备和精细化工领域具有重要的应用价值,特别是在“双碳”战略背景下,发展高产物选择性伯醇-羧酸氧化技术可以实现零碳排放的化学品制备和制氢相耦合,在规模化储能方面具有重要的应用前景。鉴于此,课题组致力于揭示镍基材料催化有机物电氧化机理,以多种伯醇(甲醇、乙醇、丙醇、丁醇、苯甲醇)氧化为羧酸为例,揭示有机物电催化氧化反应决速步骤、电子转移机制和产物选择性机理。
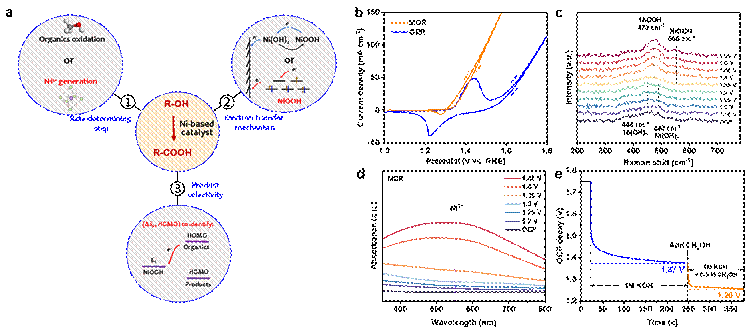
图1. 镍基材料电氧化伯醇-羧酸机理挑战。 (a) 本研究旨在理解镍基材料电氧化伯醇-羧酸反应决速步骤、电子转移机制和产物选择性机理。 (b) 甲醇氧化(红线)和水氧化(蓝线)的循环伏安曲线。 (c) 不同电压下甲醇氧化的原位电化学Raman谱。 (d) 不同电压下的甲醇氧化紫外-可见吸收谱。 (e) Ni(OH)2电极经历高电压极化后在 1 M KOH 和 0.5 M CH3OH + 1 M KOH电解液中的开路电压衰减情况。
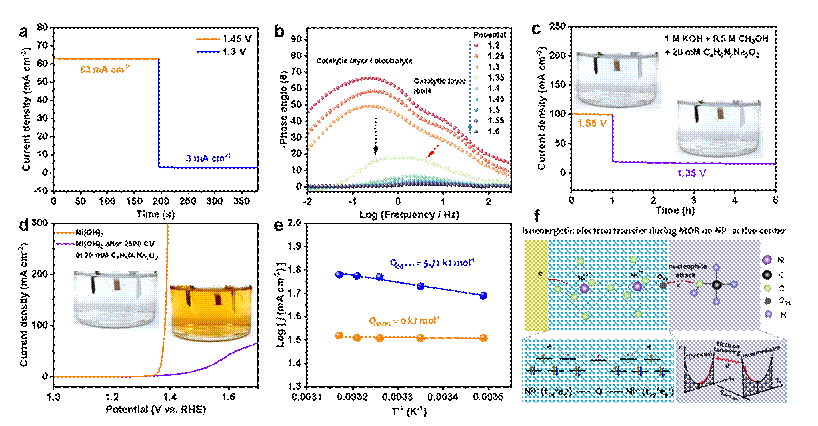
图2. Ni3+活性中心直接电子转移机制。(a)突然电压切换下的i-t曲线。(b)变电压下的电化学阻抗谱。(c)不同电压i-t条件下的Ni2+中间物种捕获,捕获剂为丁二酮肟螯合剂。插图为电解液照片,未观察到Ni2+红色络合物,说明电化学电子转移过程中不存在Ni2+中间物种。(d)Ni(OH)2电极经历了在丁二酮肟Ni2+离子螯合剂存在下2500圈循环伏安扫后的线扫伏安曲线。插图为电解液照片,红色的Ni2+红色络合物说明当电子转移过程中存在Ni2+时将被丁二酮肟络合而从电极表面剥离。(e) Ni(OH)2/NiOOH电氧化活化能和甲醇在Ni(OH)2/NiOOH氧化电压下发生电氧化时的活化能。 (f) Ni3+活性中心氧化甲醇的直接电子转移机制。当电压极化Ni(OH)2电极为NiOOH时,配不饱和的Ni3+活性位与甲醇氧化基元步物种之间建立等能电子转移通道,发生宏量界面电子隧穿经由Ni3+ (t2g6eg1) 空轨道通过Ni-O-Ni 双交换机制传输电子,不改变价态。
Ni3+生成是镍基材料电氧化伯醇-羧酸反应决速步:首先通过对比水氧化和甲醇氧化循环伏安曲线,发现当电极被电氧化从Ni(OH)2相变为NiOOH时,即可发生甲醇氧化,这意味着对于甲醇氧化反应来说,其反应决速步骤是Ni3+的产生。原位电化学Raman谱、原位电化学紫外-可见吸收谱以及开路电压衰减等研究手段确认了Ni3+是Ni(OH)2电极电氧化有机醇的活性物种。而后,针对Ni3+活性中心反应动力学开展了研究。在i-t模式下,当快速切换电压时,电化学反应能够快速建立新的平衡态,说明Ni3+氧化甲醇具有较高的反应动力学。电化学阻抗谱也证明了Ni3+氧化甲醇具有较低的界面电子转移电阻。为澄清Ni3+催化中心转移电子是否经由Ni3+/Ni2+变价过程,课题组采用丁二酮肟作为高灵敏的Ni2+络合剂(监测浓度极限nM,时间尺度ns),研究发现在Ni3+生成电压区域内快速切换电压时,电极恒定氧化甲醇并宏量转移电子,此时探测不到Ni2+。然而,在Ni2+/Ni3+氧化-还原电压范围内进行循环伏安扫的过程中,可以探测到Ni2+,这意味着在甲醇氧化过程中Ni3+转移电子并不经由Ni2+中间价态。并且Ni3+在其生成电压下氧化甲醇的活化能为0,也进一步证明了Ni3+转移电子是类导体行为。
Ni3+活性中心直接转移电子:电化学界面电子转移机制是电化学研究的重要课题。1931年,R. W. Gurney提出电化学反应电子转移量子力学机制,认为电极稳态电流起因于电极/电解液界面等能电子宏量隧穿效应。此后,1952年,W. F. Libby 提出了等能电子转移满足Franck-Condon原理,即电子转移时间尺度远快于分子构型变化。而后,在Libby研究工作基础上,R. A. Marcus对暗态化学反应如何满足能量守恒原理和Frank-Condon原理进行了深度思考,并于1955-1965年间提出了Marcus电荷转移理论(1992年诺贝尔化学奖), 明确指出电化学反应电解液可以看作是连续介电环境,反应分子核坐标波动起源于反应分子与介电环境之间以及与电极活性中心之间的相互作用,且核坐标波动是化学反应满足能量守恒原理和Frank-Condon原理并建立等能电子隧穿的基本前提。因此可以认为,外电压作用下电极催化中心被逐渐极化到等能电子转移态,同时反应分子与电解液环境和电极催化中心相互作用使得其核坐标波动而达到等能电子转移态,电化学反应因此遵循能量守恒原理和Frank-Condon原理。因此,根据Marcus电子转移原理,当电极极化Ni2+到Ni3+等能电子转移态时,甲醇分子也通过溶剂化效应及其与活性中心的相互作用而调整其几何构型达到等能电子转移态,因此克服Frank-Condon势垒(活化能)而发生等能宏量电子转移。可见,从Marcus电子转移理论角度分析,在Ni3+氧化甲醇的电子转移过程中不可能经由Ni3+/Ni2+变价过程传递电子,如果电子转移导致了Ni3+被还原为Ni2+则意味着界面能级发生了巨大的波动,此时则电子转移将严重背离Marcus电子转移机制。
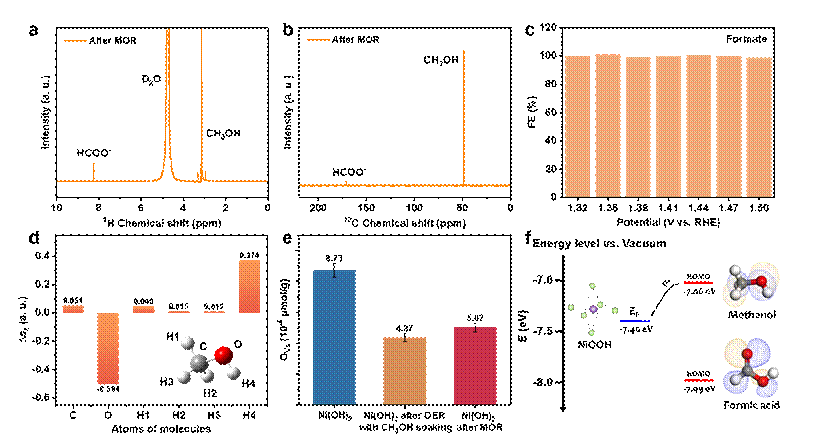
图3. Ni3+活性中心甲醇氧化产物选择性机理。(a、b)甲醇氧化产物的 1H 和 13C NMR 谱。(c)甲醇氧化产甲酸反应的法拉第效率。(d)甲醇分子中各原子的双描述符局部软度值。(e)Ni(OH)2电极在甲醇氧化后以及水氧化后浸泡甲醇的氧空位浓度变化。(f)NiOOH电极的费米能级、甲醇和甲酸的HOMO能级之间的关系。
Ni3+催化醇-羧酸产物选择性起源:1H 和 13C核磁共振谱分析显示,Ni3+催化甲醇可以单一选择性地生成甲酸,且在较宽电压范围内均展现了接近100%的法拉第效率。基于课题组前期提出的有机物电氧化反应亲核位点和分子能级热力学组合判据(Nat. Commun. 2023, 14, 7987), 对于有机亲核电氧化反应来说,利用密度泛函理论计算了分子中原子的双描述符局部软度值(dual local softness,∆sk),∆sk值的大小可以帮助判断分子中的亲核原子位,通常原子的∆sk值越负代表其成为亲核攻击位点的可能性越大。计算结果显示,甲醇分子中的羟基氧具有较负的∆sk值,意味着其是亲核攻击位点。同时,电子自旋共振谱证实了Ni(OH)2电极中存在大量配位不饱和的Ni位点(氧空位)。这意味着,甲醇分子氧化是通过羟基氧与配位不饱和的Ni3+相互作用建立等能电子转移通道。此外,对于分子氧化反应来说,其电子是经由最高占据分子轨道(HOMO)能级注入到电极的,因此分子的HOMO能级和电极费米能级之间的关系可以作为反应能否发生的热力学判据。理论计算显示,甲醇分子的HOMO能级高于NiOOH的费米能级,意味着甲醇分子HOMO能级电子化学势足够高,能够驱动电子注入NiOOH费米能级,使得甲醇分子氧化。而甲酸分子的HOMO能级则远低于NiOOH费米能级,因此得以成为产物而稳定存在。
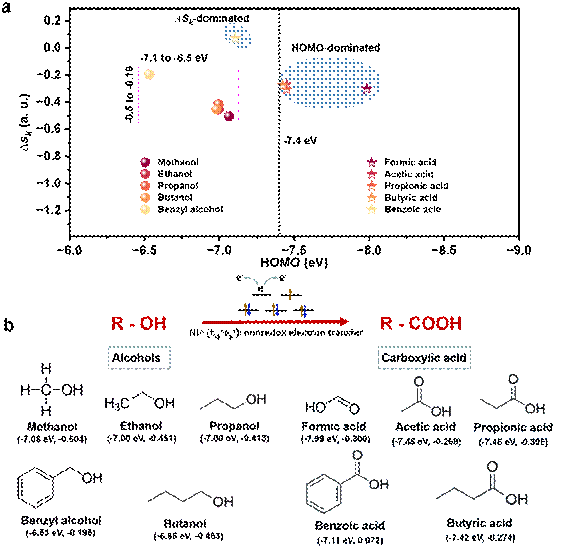
图4. 伯醇电氧化为羧酸反应的通用性。(a)分子亲核位的∆sk值和分子HOMO能级作为Ni3+氧化伯醇-羧酸反应的判据。(b)Ni3+氧化伯醇及其羧酸产物的结构式和 (HOMO, ∆sk ) 组合参数特征。
(HOMO, ∆sk)组合作为Ni3+催化醇-羧酸反应产物选择性判据:电化学实验证实,与甲醇氧化为甲酸反应类似,Ni3+可以有效氧化多种伯醇(甲醇、乙醇、丙醇、丁醇、苯甲醇)为相应的羧酸。为此,鉴于(HOMO, ∆sk)组合成功解释了甲醇-甲酸反应选择性的物理本源,我们尝试考察这一组合参数在判断多种伯醇氧化反应选择性的适用性。理论计算显示,伯醇的羟基氧均为亲核原子位,且所考察的伯醇的HOMO能级均高于NiOOH的费米能级,代表这些醇电化学氧化反应是热力学允许的过程。然而产物选择性可以划分为∆sk主控和HOMO主控两种类型:甲酸、乙酸、丙酸和丁酸的HOMO能级均深于NiOOH的费米能级,为HOMO主控类型反应;而苯甲酸的HOMO能级十分接近于NiOOH的费米能级,但其羧基氧的∆sk值为正,这是由于苯环是强吸电子集团,因此钝化了羧酸集团中的亲核原子,而使得∆sk值为正,削弱了亲核作用而使得苯甲酸稳定而作为产物存在,成为∆sk主控型反应。我们的研究证明了(HOMO, ∆sk)组合可以作为有机物电氧化发生和产物选择性的有效判据。
该工作于2024年2月7日以“Trivalent nickel-catalyzing electroconversion of alcohols to carboxylic acids”为题发表在Journal of the American Chemical Society 2024, https://doi.org/10.1021/jacs.3c13155上。37000cm威尼斯现代工程与应用科学学院博士生颜元东和硕士生钟佳颖为论文共同第一作者、闫世成教授为论文通讯作者,该研究得到了邹志刚院士的关心和指导。研究得到了国家自然科学基金和江苏省双碳专项资金资助。
文章链接 https://doi.org/10.1021/jacs.3c13155
 English
English


